Publications
Publications by categories in reversed chronological order.
2022
-
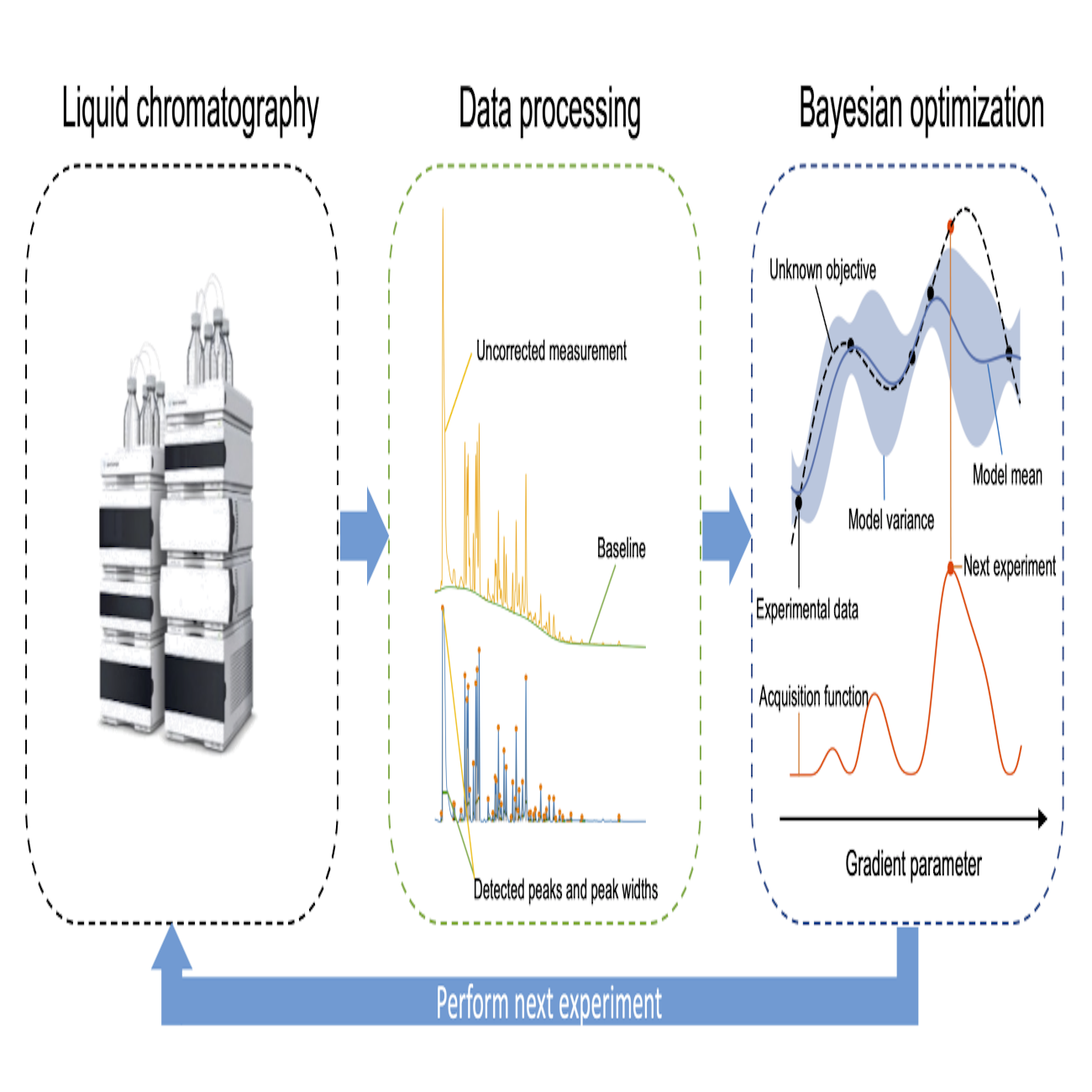 Closed-loop automatic gradient design for liquid chromatography using Bayesian optimizationBoelrijk, Jim, Ensing, Bernd, Forré, Patrick, and Pirok, BobAnalytica Chimica Acta 2022
Closed-loop automatic gradient design for liquid chromatography using Bayesian optimizationBoelrijk, Jim, Ensing, Bernd, Forré, Patrick, and Pirok, BobAnalytica Chimica Acta 2022Contemporary complex samples require sophisticated methods for full analysis. This work describes the development of a Bayesian optimization algorithm for the automated and unsupervised development of gradient programs. The algorithm was tailored to LC using a Gaussian process model with a novel covariance kernel. To facilitate unsupervised learning, the algorithm was designed to interface directly with the chromatographic system. Single-objective and multi-objective Bayesian optimization strategies were investigated for the separation of a complex (n>80) dye mixture. The multi-objective strategy was found to be very powerful and flexible in terms of exploring the Pareto front. The single-objective strategy was found to be slightly faster in finding a satisfactory optimum. One additional advantage of the multi-objective approach was that it allows a trade-off to be made between multiple objectives. In general, the Bayesian optimization strategy was found to be particularly suitable, but not limited to, cases where retention modelling is not possible, although its scalability might be limited in terms of the number of parameters that can be simultaneously optimized.
-
 Chemometric Strategies for Fully Automated Interpretive Method Development in Liquid ChromatographyBos, Tijmen, Boelrijk, Jim, Molenaar, Stef, Veer, Brian, Niezen, Leon, Herwerden, Denice, Samanipour, Saer, Stoll, Dwight, Forré, Patrick, Ensing, Bernd, Somsen, Govert, and Pirok, BobAnalytical Chemistry 2022
Chemometric Strategies for Fully Automated Interpretive Method Development in Liquid ChromatographyBos, Tijmen, Boelrijk, Jim, Molenaar, Stef, Veer, Brian, Niezen, Leon, Herwerden, Denice, Samanipour, Saer, Stoll, Dwight, Forré, Patrick, Ensing, Bernd, Somsen, Govert, and Pirok, BobAnalytical Chemistry 2022The great potential gains in separation power and analysis time that can result from rigorously optimizing LC-MS and 2D-LC-MS methods for routine measurements has prompted many scientists to develop computer-aided method-development tools. The applicability of these has been proven in numerous applications, but their proliferation is still limited. Arguably, the majority of LC methods are still developed in a conventional manner, i.e. by analysts who rely on their knowledge and experience. In this work, a novel, open-source algorithm was developed for automated and interpretive method development of LC separations. A closed-loop workflow was constructed that interacted directly with the LC and ran unsupervised in an automated fashion. The algorithm was tested using two newly designed strategies. The first utilized retention modeling, whereas the second used the Bayesian-optimization machine-learning approach. In both cases, the algorithm could arrive within ten iterations at an optimum of the objective function, which included resolution and measurement time. The design of the algorithm was modular, so as to facilitate compatibility with previous works in literature and its performance thus hinged on each module (e.g., signal processing, choice of retention model, objective function). Key focus areas for further improvement were identified. Bayesian optimization did not require any peak tracking or retention modeling. Accurate prediction of elution profiles was found to be indispensable for the strategy using retention modeling. This is the first interpretive algorithm demonstrated with complex samples. Peak tracking was conducted using UV-Vis absorbance detection, but use of MS detection is expected to significantly broaden the applicability of the workflow.
-
 Predicting RP-LC retention indices of structurally unknown chemicals from mass spectrometry dataBoelrijk, Jim, Herwerden, Denice, Samanipour, Saer, Ensing, Bernd, and Forré, PatrickJournal of Cheminformatics 2022
Predicting RP-LC retention indices of structurally unknown chemicals from mass spectrometry dataBoelrijk, Jim, Herwerden, Denice, Samanipour, Saer, Ensing, Bernd, and Forré, PatrickJournal of Cheminformatics 2022Non-target analysis combined with high resolution mass spectrometry is considered one of the most comprehensive strategies for the detection and identification of known and unknown chemicals in complex samples. However, many compounds remain unidentified due to data complexity and limited structures in chemical databases. In this work, we have developed and validated a novel machine learning algorithm to predict the retention index (r_i) values for structurally (un)known chemicals based on their measured fragmentation pattern. The developed model, for the first time, enabled the predication of r_i values without the need for the exact structure of the chemicals, with an R^2 of 0.91 and 0.77 and root mean squared error (RMSE) of 47 and 67 r_i units for the Norman and amide test set, respectively. This fragment based model showed comparable accuracy in r_i prediction compared to conventional descriptor-based models that rely on known chemical structure, which obtained a R^2 of 0.85 with and RMSE of 67.
2021
-
 Bayesian optimization of comprehensive two-dimensional liquid chromatography separationsBoelrijk, Jim, Pirok, Bob, Ensing, Bernd, and Forré, PatrickJournal of Chromatography A 2021
Bayesian optimization of comprehensive two-dimensional liquid chromatography separationsBoelrijk, Jim, Pirok, Bob, Ensing, Bernd, and Forré, PatrickJournal of Chromatography A 2021Comprehensive two-dimensional liquid chromatography (LCxLC), is a powerful, emerging separation technique in analytical chemistry. However, as many instrumental parameters need to be tuned, the technique is troubled by lengthy method development. To speed up this process, we applied a Bayesian optimization algorithm. The algorithm can optimize LCxLC method parameters by maximizing a novel chromatographic response function based on the concept of connected components of a graph. The algorithm was benchmarked against a grid search (11,664 experiments) and a random search algorithm on the optimization of eight gradient parameters for four different samples of 50 compounds. The worst-case performance of the algorithm was investigated by repeating the optimization loop for 100 experiments with random starting experiments and seeds. Given an optimization budget of 100 experiments, the Bayesian optimization algorithm generally outperformed the random search and often improved upon the grid search. Moreover, the Bayesian optimization algorithm offered a considerably more sample-efficient alternative to grid searches, as it found similar optima to the grid search in far fewer experiments (a factor of 16–100 times less). This could likely be further improved by a more informed choice of the initialization experiments, which could be provided by the analyst’s experience or smarter selection procedures. The algorithm allows for expansion to other method parameters (e.g., temperature, flow rate, etc.) and unlocks closed-loop automated method development.
2019
-
 Incorporating maximally localized Wannier functions into neural network potentials [Master thesis, supervised by Ambuj Tiwari]Boelrijk, Jim2019
Incorporating maximally localized Wannier functions into neural network potentials [Master thesis, supervised by Ambuj Tiwari]Boelrijk, Jim2019A efficient and accurate description of the inter-atomic potential energy surface (PES) is immensely important for molecular modelling, which is a fundamental tool for chemistry, computational biology and material science. While electronic structure methods are accurate, they are computationally expensive. Approximation methods typically are more computationally efficient, yet this comes with a reduction in accuracy. In recent years, the development of atomistic potentials using machine learning (ML) techniques have offered a solution to the accuracy versus efficiency problem. A type of ML, deep neural networks, can "learn" the topology of the PES by adjusting a number of parameters with the aim to reproduce a set of reference \textitab initio data as accurately as possible. These neural network potentials (NNPs) have been used to describe chemical systems, ranging from small organic molecules to complex alloys with high accuracy compared to the underlying electronic structure method, at a fraction of the computational cost. In addition, NNPs allow for linear scaling with system size, making them both accurate and efficient for the study of larger system. Their flexible construction also allows for the making and breaking of bonds, which enables reactivity studies. However, current NNP methods are purely atomistic, and although trained on electronic structure methods, do not incorporate electronic information directly, which limits their application to the computation of non-electronic properties. This thesis, investigated the incorporation of electronic information, in the form of maximally localised Wannier functions (MLWFs), within existing NNP frameworks in order to improve accuracy and transferability, while also allowing for the analysis of electronic information. Two scenarios where implemented in the DeepPot-SE method. In the first scenario, the Wannier function centers (WFCs) were considered as classical particles and incorporated in the NNP structure as well as in the local input. In scenario 2, the WFCs were only incorporated in the local input. Although scenario 1 resulted in high-accuracy predictions of the data set, it was not usable for molecular dynamics simulations. It was not evident what mass the WFCs should be given for proper propagation. In addition, a more complete data set must be used in order to cover the state-space of the WFCs, necessary for this scenario. Scenario 2 did not improve prediction accuracy over the conventional methods and a incorporation of the spreads of the WFCs might be necessary to improve this.